Scripts
-- last updated 31 October 2006 --
UNDER CONSTRUCTION
McMahon Lab Home
GenBank to
supermatrices pipeline
To study the potential of the sequence data in GenBank to resolve a tree for a clade of interest to my research, the Papilionoid legumes, I developed a "pipeline" in collaboration with Mike Sanderson and the Phylota project. See paper (McMahon and Sanderson, 2006, Phylogenetic supermatrix analysis of GenBank Sequences from 2228 Papilionoid Legumes, Systematic Biology 55:818-836) for a full description of the methods for this project.
A few of the many scripts involved are available here; contact me for additional tools mentioned in the paper.
| blast2blink.pl Download current version: v. 0.37 This tool is used for controlling the levels of length heterogeneity when building single-linkage clusters of sequences. BLAST's table output (achieved using the command -m 8) is parsed and compared to a table that contains lengths for each of the sequences. Used in combination with blink (M. J. Sanderson, written in C) or blinkPerl (below), it is similar in conception to NCBI's BLASTClust. However, the separation of the blasting and the clustering steps allows full use of blast's various programs (blastn, etc.), and a critical difference (as of this writing) is that blastclust considers one hit at a time when calculating proportional overlap, whereas blast2blink considers the entire set of hits between a pair of sequences. This means that a pair of sequences with, e.g., a string of N's or other low-complexity runs in the middle can still be considered a hit if the surrounding regions are found to be similar enough. 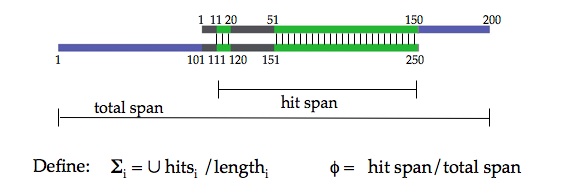 Running the program with no commands produces detailed instructions: ./blast2blink.pl After downloading the script, it may be necessary to make it executable: chmod +x blast2blink.pl The script has been tested with a wide set of parameter values and commands, but not exhaustively, so it is still in Beta form. If errors occur, please contact me. NOTE: a bug in v. 0.35 has been fixed, and the instructions have been updated. |
| blinkPerl.pl Download current version: v. 1.0 This tool is used for making clusters of sequences. Input consists of pairs of sequence identifiers that "hit" to each other, output is a list of clusters and their consituent sequences. A sequence is added to a cluster if it hits to any other sequence in the cluster (single-linkage clustering). Running the program with no commands produces a list of commands, read the source for formatting details. Please contact me with any errors or problems. After downloading the script, it may be necessary to make it executable: chmod +x blinkPerl.pl |