List
of the software/R packages developed in my lab:
|
ENNB: |
A two-stage statistical procedure for
feature selection and comparison in functional analysis of metagenomes |
|
metaFunction: |
R package for
statistical profiling functions in a microbial community |
|
FunctionSIM: |
(Java
software) A sequencing simulator for functional metagenomics |
|
TAEC: |
R package
for Taxonomic Analysis by Elimination and Correction on closely related
species |
|
TAMER: |
R package for accurate taxonomic assignment of metagenomic sequencing reads |
ENNB: A
two-stage statistical procedure for feature selection and comparison in
functional analysis of metagenomes:
In
the first stage of the proposed procedure, the informative features are
selected using Elastic Net as
reducing the dimension of metagenomic data; in the
second stage the differentially abundant features are detected using
generalized linear models with a Negative
Binomial distribution.
This
is an introduction (README
file)
to using the R code for the proposed method for detection of significantly
differentially abundant features of different metagenomic
communities/conditions.
Example data
for two-group comparison (feature
count and phenotype
info)
Example data
for multiple-group comparison (feature
count and phenotype
info)
Citation: Pookhao N, Sohn M, Li Q, Jenkins I, Du R, Jiang H, An L.
(2014) A two-stage statistical procedure for feature selection and comparison
in functional analysis of metagenomes. Bioinformatics (in
press).
metaFunction: A statistical
tool in profiling functions in a microbial community
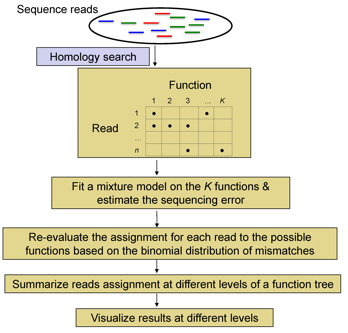
Flowchart of metaFunction
Download the metaFunction package and the associated manual
file, and the simulated
sequence data (.fasta) in the metaFunction
paper.
More details can be found here.
Citation: An L, Pookhao N, Jiang H, Xu J. (2014) Statistical approach of functional profiling
for a microbial community. PLoS ONE 9(9): e106588.
doi:10.1371/journal.pone.0106588
FunctionSIM: A sequencing simulator for functional metagenomics
As standalone software it allows users to simulate metagenomic sequence datasets that can be used as standardized
test data for planning metagenomic projects or for
benchmarking software in functional metagenomic
analysis.
More details can be found at here.
TAEC: an R
package for Taxonomic Analysis by Elimination and Correction
TAEC, a new
homology-based approach for taxonomic analysis, utilizes the similarity in the
genomic sequence in addition to the result of an alignment tool.
This approach
consists of two main stages: the elimination stage and the correction stage. In
the elimination stage, the potential true genomes identified by removing false
genomes whose presence is most likely due to the presence of similar genomes in
a sample. In the correction stage, the abundances of the genomes remaining
after the elimination stage are corrected by utilizing the similarity between
genomes in a system of linear equations. The overall workflow of TAEC is shown
as below.
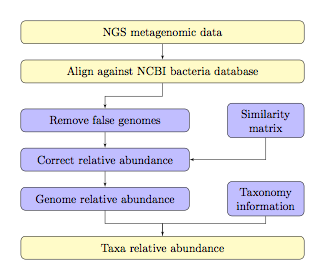
Note:
¾ The light yellow colored blocks
are implemented by a user and the light blue colored blocks are internally implemented by TAEC.
¾ The bacteria database could be replaced with virus
or other types of databases if needed.
¾ Similarity matrix is given for different lengths of
sequence reads, at 100pb, 250pb, 500pb, and 1000bp.
R package of TAEC can be downloaded: (Mac version)
and (linux version)
Note:
We have tested the TAEC package on R
version 2.14.1 (2011-12-22) and version 2.15.2 (2012-10-26) on Redhat and R version 3.0.2 (2013-09-25) on Ubuntu. No
errors associated with the different versions of R occurred. On the other hand,
we encountered errors associated with different versions of R on Mac OSX. In
order for the TAEC package to work properly on OSX, please upgrade your R to
the current version of 3.0.2 (2013-09-25).
Citation: Sohn M, An L, Pookhao N, Li Q. (2014) Accurate genome relative abundance
estimation for closely related species in a metagenomic
sample. BMC Bioinformatics 15:242.
TAMER: an R package for accurate taxonomic assignment of metagenomic
sequencing reads.
This is a
collaborative work with Hongmei Jiang’s lab.
More
details can be found at here.
Citation: Jiang H *, An L*, Lin
SM, Feng G, Qiu Y. (2012) A
Statistical Framework for Accurate Taxonomic Assignment of Metagenomic
Sequencing Reads. PLoS ONE 7(10): e46450.
(*: co-first author)